

X射线衍射仪原理——X射线衍射法 发布日期:2014-02-02 00:00:00 文章来源:莱雷科技
X射线衍射法是一种利用单色X射线光束照射到一颗晶体(单晶X射线衍射法,SXRD)或众多随机取向的微小晶体(粉末X射线衍射法,PXRD)来测量样品分子三维立体结构或特征X射线衍射图谱的检测分析方法。①
固体化学物质状态可分为晶态(或称晶体)和非晶态(或称无定型态、玻璃体等)物质两大类。
晶态物质(晶体)中的分子、原子或离子在三维空间呈周期性有序排列,晶体的最小重复单位是晶胞。晶胞是由一个平行六面体组成,含有三个轴(a、b、c,单位:Å)和三个角(α、β、γ,单位:°),被称为晶胞参数。晶胞沿(x、y、z)三维的无限有序堆积排列形成了晶体。
非晶态物质(无定型态、玻璃体等)中的分子、原子或离子在三维空间不具有周期性排列规律,其固体物质是由分子、原子或离子在三维空间杂乱无章的堆积而成。
X射线衍射的基本原理:当一束X射线通过滤波镜以单色光(特定波长)照射到单晶体样品或粉末微晶样品时即发生衍射现象,衍射条件遵循布拉格方程式:
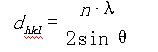
式中 dhk
为面间距(hkl为晶面指数);
n为衍射级数;
λ为X射线的波长;
θ为掠射角。
金属铜(Cu)与钼(Mo)为有机化合物样品常用的X射线阳极靶元素,Cu靶波长λ为1.54178Å,Mo靶波长λ为0.71073Å。X射线由Kα和Kβ组成,一般采用Kα线作为单晶X射线衍射的结构分析或粉末X射线衍射的成分与晶型分析的特征X射线。②[2]
当X射线照射到晶态物质上时,可以产生衍射效应;而当X射线照射到非晶态物质上时则无衍射效应。单晶X射线衍射结构(晶型)定量分析和粉末X射线成分(晶型)定性与定量分析均是依据X射线衍射基本原理。
X射线衍射仪器是由X射线光源(直流高压电源、真空管、阳极靶)、准直系统(准直管、样品架)、仪器控制系统(指令控制、数据控制)、冷却系统组成。
一、单晶X射线衍射法 ③
单晶X射线衍射分析法使用一颗单晶体即可获得样品的化合物分子构型和构象等立体结构信息,主要包括:空间群、晶胞参数、分子式、结构式、原子坐标、成键原子的键长与键角、分子内与分子间的氢键、盐键、配位键等。
单晶X射线衍射分析技术是定量检测样品成分与分子立体结构的绝对分析方法,它可独立完成对样品的手性或立体异构体分析、共晶物质分析(含结晶水或结晶溶剂等)、纯晶型物质分析(分子排列规律变化)等。由于单晶射线衍射分析实验使用一颗晶体,所以采用该分析法可获得晶型纯品物质信息。
单晶X射线衍射分析通过两次傅里叶变换完成结构分析目的。该方法适用于对晶态物质的结构或晶型分析。单晶X射线衍射实验中,Cu靶适用对化合物分子的绝对构型测定, Mo靶适用对化合物分子的相对构型测定(含有卤素或金属原子的样品除外)。
试样的制备及有关实验技术:
试样制备:单晶X射线衍射分析要求使用一颗适合实验的单晶体,一般需要采用重结晶技术通过单晶体培养获得。晶体尺寸在0.1~1.0mm之间。单晶体应呈透明状、无气泡、无裂纹、无杂质等,晶体外形可为块状、片状、柱状,近似球状或块状晶体因在各方向对X射线的吸收相近,所以属最佳实验用晶体外形。
晶体样品对X射线的衍射能力受到来自内部和外部的影响。晶体样品自身内部影响因素主要为组成晶体的化学元素种类、结构类型、分子对称排列规律、作用力分布、单晶体质量等;仪器外部影响因素包括X射线发生器功率、阳极靶种类等。
当使用Cu靶实验时,衍射数据收集的2θ角要大于114°;当使用Mo靶实验时,衍射数据的收集的2θ角要大于54°。
晶胞参数三个轴(a、b、c,单位:Å)的误差在小数点后第三位,三个角(α、β、γ,单位:°)的误差在小数点后第二位;原子相对坐标的误差在小数点后第四位,键长的误差在小数点后第三位,键角的误差在小数点后第一位。
本法适用于样品的定量分子立体结构分析、手性分析、晶型分析、结晶水含量分析、结晶溶剂种类与含量分析等。
仪器校准:仪器应定期使用仪器生产厂家自带的标准样品进行仪器校正。
二、粉末X射线衍射法
粉末X射线衍射法可用于样品的定性或定量的物相分析。每种化学物质,当其化学成分与固体物质状态(晶型)确定时,应该具有独立的特征X射线衍射图谱和数据,衍射图谱信息包括衍射峰数量、衍射峰位置(2θ值或d值)、衍射峰强度(相对强度,绝对强度)、衍射峰几何拓扑(不同衍射峰间的比例)等。④
粉末X射线衍射法适用于对晶态物质或非晶态物质的定性鉴别与定量分析。常用于固体物质的结晶度定性检查、多晶型物质种类、晶型纯度等分析。粉末X射线衍射实验中,通常使用Cu靶为阳极靶材料。
晶态物质的粉末X射线衍射峰是由数十乃至上百个锐峰(窄峰)组成;而非晶态物质的粉末X射线衍射峰的数量较少且呈弥散状(为宽峰或馒头峰),在定量检测时,二者在相同位置的衍射峰的绝对强度值存在较大差异。
当化学物质有两种或两种以上的不同固体物质状态时,即存在有多晶型(或称为同质异晶)现象。多晶型现象可以由样品的分子构型、分子构象、分子排列规律、分子作用力等变化引起,也可由结晶水或结晶溶剂的加入(数量与种类)形成。每种晶型物质应具有确定的特征粉末X射线衍射图谱。
当被测定样品化学结构相同,但衍射峰的数量和位置、绝对强度值或衍射峰形几何拓扑间存在差别时,即表明该化合物可能存在多晶型现象。
试样的制备及有关实验技术:
试样的制备:粉末晶体颗粒过大或晶体呈现片或针状样品容易引起择优取向现象,为排除择优取向对实验结果的干扰,对有机样品需要增加研磨并过筛(通常为100目)的样品前处理步骤。
实验进样量:当采用粉末X射线衍射技术进行定量分析时,需要对研磨后过筛样品进行精密定量称取,试样铺板高度应与板面平行。
衍射数据收集范围:当使用铜Cu靶实验时,衍射数据收集的范围(2θ)一般至少应在④ 增加了粉末X射线衍射法获得的衍射图谱的信息,并对该法的适用范围进行了描述。3~60°之间,有时可收集至1~80° ⑤。
定量分析方法:可采用标准曲线法,含外标法、内标法与标准加入法⑥。
定量分析时,应选择一个具有特征性的衍射峰进行。内标法应建立内标物质与衍射强度之间的线性关系。内标物质选取原则是应与样品的特征衍射峰不发生重叠,同时二者对X射线的衍射能力应接近。制作标准曲线时,应取固定质量但含量比例不等的内标物质与样品均匀混合,定量分析时,应保证被测样品含量在标准曲线的线性范围内;外标法应建立标准物质不同质量与衍射强度之间的线性关系。制作标准曲线时,应取不同质量的样品。定量分析时,应保证被测样品含量在标准曲线的线性范围内;标准加入法应保证加入标准物质和被测物质衍射峰强度接近,二者具有良好的分离度且不重叠。
定量分析时,应平行实验3次,取算术平均值。当样品存在多晶型物质状态,且压力能引起晶型转变时,定量分析方法应慎用。当多晶型衍射图谱的衍射峰数量和位置基本相同,但衍射峰的几何拓扑图形存在较大差异时,应适当增加特征衍射峰的数量(从一般使用1个特征峰,增加到使用3~5个特征峰),以证明晶型含量与特征衍射峰间存在线性关系。
采用相同制备方法的等质量试样定量分析,在同一实验条件下,样品与标准品的2θ值数据误差范围为±0.2°,衍射峰的相对强度误差范围为 ±5%,否则应考虑重新进行实验或可能存在多晶型问题。⑦
本法适用于样品的结晶性检查、样品与标准品的异同性检查、样品生产工艺稳定性检查、样品化学纯度检查与定量分析(当杂质成分含量大于1% 时在衍射图谱中可以识别),样品的晶型鉴别与晶型纯度定量分析等。
仪器校准:仪器应定期使用标准物质Al2 O3或单晶硅粉进行仪器校正。⑧
起草单位:医科院药物研究所
审校:医科院药物研究所 吕扬
①X射线衍射法分为单晶X射线衍射法与粉末X射线衍射法,两种分析方法具有不同的应用范围,故在新版药典中将X射线粉末衍射法修订为X射线衍射法,并收录了单晶X射线衍射分析方法与粉末X射线衍射分析方法。新修订的X射线衍射法对原理及晶体学表述进行了规范,使之更符合药物晶体学术语及表达习惯,使其更具科学性和准确性。
② 有机化学药物分析常用金属靶元素与波长。
③ 增加了单晶X射线衍射分析法相关内容。
⑤当使用Cu靶实验时,衍射数据收集的范围(2θ)应在3~80°,因为一些有机药物分子通常在60°以后仍然存在大量的衍射峰。
⑥ 外标标准曲线法亦是常用的定量分析方法,应予以介绍。
⑦ 定量分析时,需要准确测定衍射强度,故在实验中应采取平行试验以消除偶然误差,并严格规定数据的误差范围(衍射峰的相对强度误差范围为从±20%修订为±5%),以获得准确的测量结果。
⑧ 增加仪器校准部分,以保证获得数据的准确性。

